


 扫码安装网站APP(Android版)
扫码安装网站APP(Android版)
来源公众号:见色非色
分子生物学 · 技术革命
PCR技术:为什么它改变了我们读取生命密码的方式
一项从试管里的手工操作,变成全球实验室标配的技术,究竟做对了什么?
1983年,美国生物化学家凯利·穆利斯(Kary Mullis)在Cetus公司工作期间,开车经过加州101号公路时突然想到一个念头:如果让DNA在体外反复复制,不就能从极微量的样本里”放大”出足够多的目标片段吗?三年后,这个想法变成了论文;十年后,他因此获得诺贝尔化学奖。而这项技术——聚合酶链式反应(PCR)——至今仍是分子生物学实验室里最基础、最不可或缺的工具之一。
一个看似简单的想法,为什么花了十几年才落地?
DNA复制的原理,早在1953年沃森和克里克提出双螺旋结构时就已隐约可见。1958年,梅塞尔森和斯塔尔用实验证实了DNA的半保留复制模型。到1971年,霍拉纳(Khorana)甚至已经提出了”体外核酸扩增”的设想。但PCR真正诞生,却是在十二年后的1983年。
问题出在一个关键环节:酶。
DNA复制需要DNA聚合酶来催化。但当时的聚合酶——比如大肠杆菌来源的Klenow片段——在90°C以上的高温中会彻底失活。而PCR的每一步循环都需要先把双链DNA加热到95°C左右使其”变性”解开。这意味着,每完成一轮循环,实验人员就得手动补加一次新鲜的酶。一个典型的PCR反应要跑30到40轮,也就是说,科研人员得在四五个小时里反复开盖、加酶、换水浴,既繁琐又容易污染。
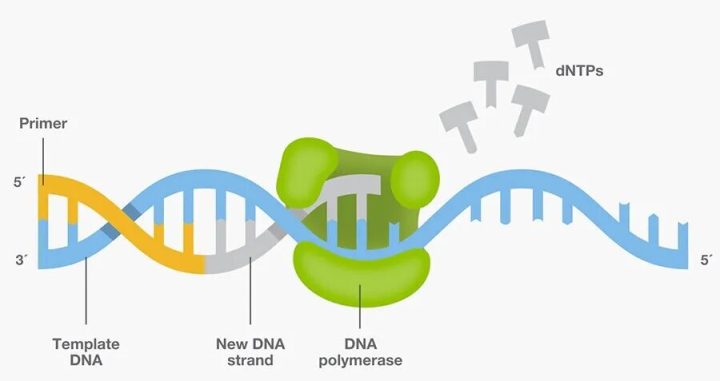
PCR反应的核心:引物引导DNA聚合酶在模板链上合成互补新链
转机出现在1976年。科学家从黄石国家公园的热泉中分离出一种嗜热细菌——水生栖热菌(Thermus aquaticus)。这种细菌生活在70°C以上的高温环境中,它的DNA聚合酶(后来被命名为Taq聚合酶)自然演化出了耐高温的特性。1988年,赛基(Randal Saiki)团队将Taq酶引入PCR体系,终于解决了”每轮加酶”的痛点。
同年,商业化热循环仪问世。PCR从此从手工艺术变成了可标准化、可重复的工业流程。1989年,《科学》杂志将DNA聚合酶分子评为”年度分子”——这大概是科学界对一种蛋白质最隆重的致敬。
1953
沃森与克里克提出DNA双螺旋结构,暗示了自我复制的可能机制。
1971
霍拉纳首次提出体外核酸扩增的理论设想,但缺乏耐热酶,无法实践。
1976
从黄石热泉中分离出水生栖热菌,其DNA聚合酶耐高温的特性为PCR自动化埋下伏笔。
1983
穆利斯在Cetus公司提出PCR核心概念:利用双引物实现DNA片段的指数级扩增。
1988
Taq聚合酶被引入PCR体系;首台商业化热循环仪问世,技术实现自动化。
1993
穆利斯因发明PCR技术获得诺贝尔化学奖。
PCR到底在做什么?三个温度,一场指数级复制
PCR的本质,是在一根小小的PCR管里,用温度变化来指挥一场精密的分子舞蹈。整个过程只需要三种温度,循环30到40次,就能把一段特定的DNA从1个拷贝放大到10亿个拷贝。
2^30 ≈ 10^9
30轮循环后,目标DNA片段的理论扩增倍数
第一步:变性(95°C)——双链DNA像拉链一样被高温”撕开”,变成两条单链。这一步摧毁了双链之间的氢键,但不会破坏磷酸骨架。
第二步:退火(50-65°C)——温度降下来,人工合成的两段短DNA(引物)找到各自互补的位置,像磁铁一样吸附上去。引物的长度通常在18到25个碱基之间,它们决定了扩增的”起点”和”终点”,也决定了PCR的特异性——只有与引物匹配的目标序列才会被复制。
第三步:延伸(72°C)——Taq聚合酶以单链DNA为模板,从引物的3’端开始,逐个添加与模板互补的碱基(dNTP),合成出一条全新的互补链。Taq酶的最适工作温度恰好是72°C左右。
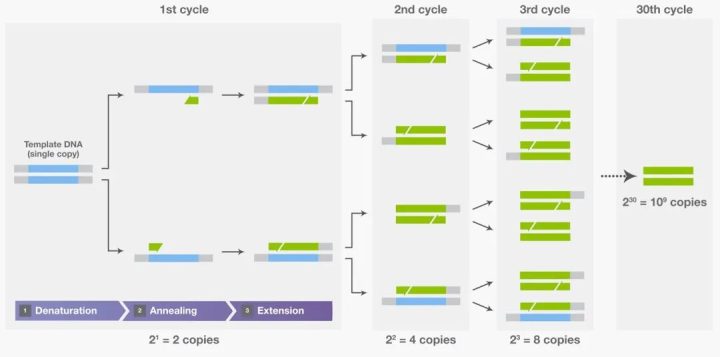
PCR的指数级扩增:每一轮循环,目标片段的数量都翻倍
三轮循环过后,原本只有1个拷贝的目标片段变成了8个。到第30轮,理论上是10亿个。当然,现实中会受到酶活性衰减、底物耗尽、引物二聚体等因素的限制,实际产量通常低于理论值。但即便如此,从一根头发丝上的微量DNA,到足以检测分析的样本量,PCR只需要两三个小时。
为什么PCR成了现代生物学的”基础设施”?
PCR的价值,不在于它本身有多复杂,而在于它解决了一个根本性的瓶颈:DNA太少了。
在PCR出现之前,研究一段特定的基因序列,通常需要先从大量细胞中提取DNA,然后用限制性内切酶切割,再经过克隆、筛选、测序——整个过程可能需要数周甚至数月。而且,如果样本中目标DNA的含量极低(比如犯罪现场的一根毛发、一滩血迹,或者古代化石中的残留物),传统方法几乎无能为力。
PCR改变了这个局面。它让”微量”不再等于”无法检测”。
PCR的灵敏度有多高?
理论上,PCR可以从单个DNA分子开始扩增。在实际应用中,法医DNA鉴定常常从犯罪现场提取到纳克(ng,十亿分之一克)级别的DNA样本,通过PCR扩增后,足以完成个体识别。新冠疫情期间,核酸检测的核心步骤也是PCR——从鼻咽拭子采集到的极微量病毒RNA,先逆转录成DNA,再通过PCR放大到可检测的水平。
除了灵敏度,PCR的另一个关键特性是特异性。引物就像两把精准的钥匙,只有与目标序列完全匹配的位置才会被”打开”。这意味着,在复杂的基因组背景中,PCR可以只复制你感兴趣的那一段DNA,而忽略其他99.99%的序列。
从PCR到qPCR、dPCR:技术如何自我进化?
最初的PCR只能回答”有”或”没有”——目标序列是否存在。但科学家很快发现,如果能实时监测扩增过程,就能回答”有多少”。
实时荧光定量PCR(qPCR)在反应体系中加入了荧光探针。每合成一条新链,探针就会释放一个荧光信号。荧光强度与DNA拷贝数成正比,通过标准曲线可以精确计算出样本中初始的DNA含量。这在基因表达分析、病原体载量检测、肿瘤标志物定量等领域至关重要。
第三代技术——数字PCR(dPCR)——则走了另一条路。它将样本分割成数万个微反应单元,每个单元要么有目标DNA(阳性),要么没有(阴性)。通过统计阳性单元的比例,可以直接计算出原始样本中的DNA浓度,无需标准曲线,也无需担心扩增效率的差异。数字PCR的灵敏度比qPCR又提高了一个数量级,特别适合检测低频突变或微量病原体。
| 技术代际 | 核心能力 | 典型应用 |
|---|---|---|
| 第一代 PCR | 定性检测(有/无) | 基因克隆、法医鉴定、病原体筛查 |
| 第二代 qPCR | 实时定量(有多少) | 基因表达分析、病毒载量监测、肿瘤标志物 |
| 第三代 dPCR | 绝对定量(精确数) | 低频突变检测、液体活检、微量病原体 |
PCR的边界:它不能做什么?
PCR不是万能的。它的第一个局限是长度。Taq聚合酶的”持续合成能力”(processivity)有限,通常一次只能合成几千个碱基。如果要扩增更长的片段,需要用到特殊的高保真聚合酶或长片段PCR技术。
第二个局限是保真度。Taq酶在合成新链时,大约每10万个碱基会引入一个错误。对于克隆和测序来说,这个错误率太高。因此,高保真PCR需要使用具有3’→5’外切酶活性的聚合酶(如Pfu酶),它们可以在合成过程中”校对”错误,将错误率降低到百万分之一以下。
第三个局限是引物设计。PCR的特异性完全依赖于引物。如果引物与非目标序列存在部分匹配,就可能产生非特异性扩增产物。引物之间如果互补,还会形成”引物二聚体”,消耗反应资源,干扰结果。设计一对好的引物,本身就是一门手艺。
PCR技术的发明,建立在DNA双螺旋结构、半保留复制机制、耐热酶发现等多个科学突破之上。它不是某个天才的灵光一现,而是几十年知识积累的必然产物。穆利斯自己也曾说过,PCR的想法”来得太容易了”——容易到让人怀疑它是否真的能工作。但正是这种”简单”,让PCR成为了现代生物学最基础、最普及的工具之一。
写在最后:一项技术如何改变一个学科
PCR的故事告诉我们,一项技术的真正价值,不在于它本身有多精妙,而在于它解决了什么级别的瓶颈。在PCR之前,DNA分析受限于样本量和操作复杂度;在PCR之后,问题变成了”你想检测什么”,而不是”你有没有足够的DNA”。
从法医DNA鉴定到新冠病毒检测,从基因诊断到考古遗传学,PCR已经渗透进了现代生命科学的每一个角落。它让”读取生命密码”从少数顶尖实验室的特权,变成了普通科研人员日常工作的一部分。这或许就是穆利斯在1983年那个加州公路上的瞬间,所真正开启的东西。



近期评论